Sindrom Maroteaux-Lamy atau Mucopolysaccharidosis VI adalah penyakit yang jarang diwarisi, di mana para pengangkut mempunyai ciri-ciri berikut:
- Sikap pendek,
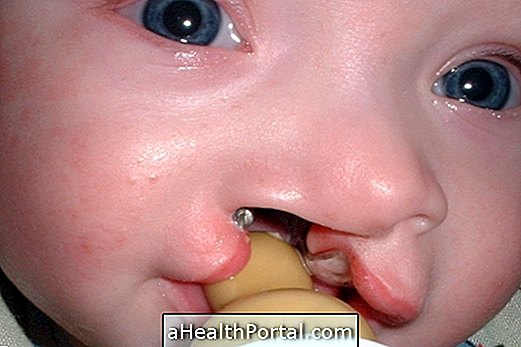
- ubah bentuk muka,
- leher pendek,
- otitis berulang,
- penyakit di saluran udara,
- malformasi rangka dan
- kekejangan otot.
Penyakit ini disebabkan oleh perubahan dalam enzim Arylsulfatase B, yang menghalangnya daripada melaksanakan fungsinya iaitu merendahkan polisakarida, yang seterusnya terkumpul di dalam sel, membangunkan ciri-ciri gejala penyakit.
Pesakit dengan sindrom mempunyai kecerdasan biasa, jadi anak-anak tidak memerlukan sekolah khas, hanya menyesuaikan bahan yang memudahkan interaksi dengan guru dan teman sekelas.
Diagnosis dibuat oleh ahli genetik berdasarkan penilaian klinikal dan analisis biokimia makmal. Diagnosis pada tahun-tahun pertama kehidupan adalah sangat penting untuk menghuraikan rancangan intervensi awal, yang akan membantu dalam perkembangan anak dan dalam rujukan ibu bapa kepada kaunseling genetik, kerana mereka menjalankan risiko melewati penyakit ini kepada anak-anak kemudian.
Tiada ubat untuk sindrom Maroteaux-Lamy, tetapi beberapa rawatan seperti pemindahan tulang sumsum dan terapi penggantian enzim membuktikan berkesan dalam mengurangkan gejala. Terapi fizikal digunakan untuk mengurangkan kekejangan otot dan membesarkan pergerakan badan seseorang. Tidak semua pesakit hadir semua gejala penyakit, keparahan berbeza dari individu ke individu, ada yang dapat menjalani kehidupan yang agak normal.